A Beginners Guide to using Balena
How to log on to, transfer data, and run jobs on the HPC at Bath
This informal guide is for people new to using Balena and effectively want to take code they have written to run on their laptop in something like Python or MATLAB and now run it on Balena. This is by no means exhaustive and there is much more information on the wiki page. In fact, much of the information on Balena here is just condensed from that website. The rest of this guide is general tips on how to transfer files and navigate the environment.
Logging onto Balena
Purely from a terminal
If you are on campus you can skip the first step and ssh directly into balena. Otherwise open a terminal and type
ssh user123@linux.bath.ac.uk
This opens up a link to the uni server (ssh stands for secure shell). Put in your password and then do the same again but change linux to balena:
ssh user123@balena.bath.ac.uk
Great, you are now in your personal home directory on balena.
Kitty
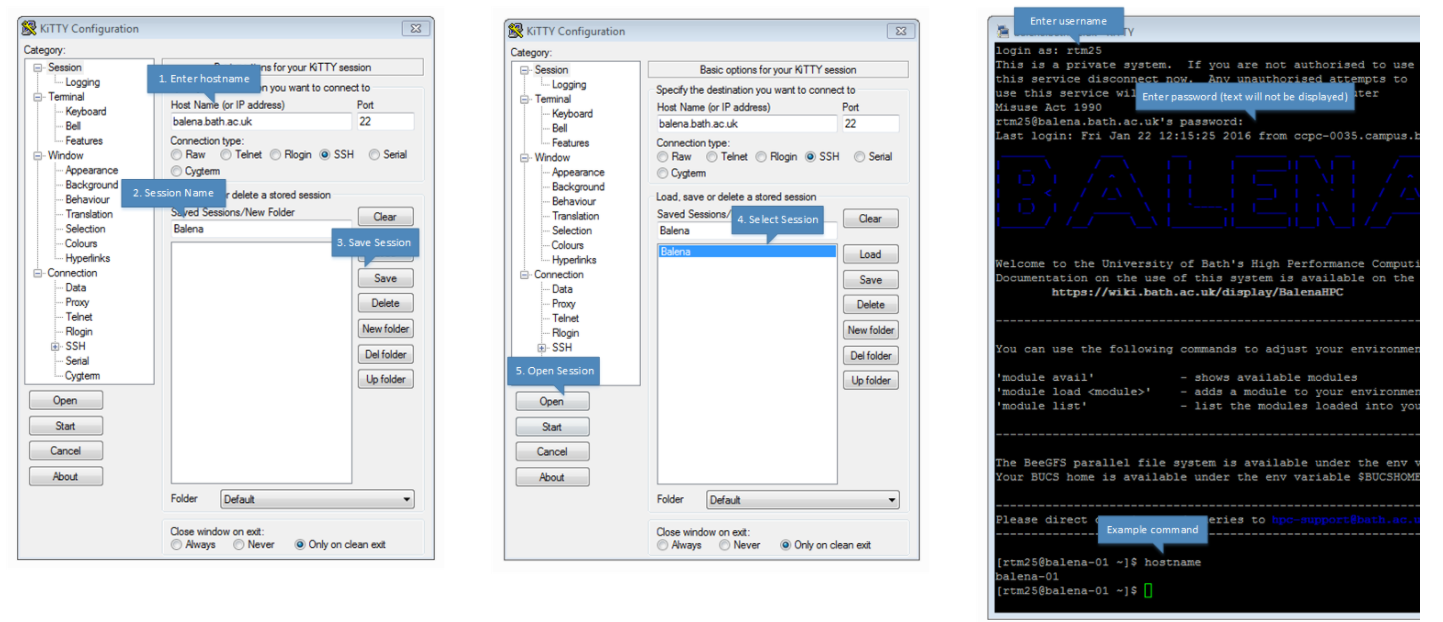
Using the image below taken from the wiki page should be sufficient in explaining how to login using Kitty.
Transferring data or files
Since you are running your code on balena, you will at some point have to move around data using a terminal so I would recommend doing this for all the steps. The added benefit of this is that your commands are stored so you can always just reuse your previous commands using the up arrows if you can’t remember exactly what a command should be.
To move files between your PC and balena you will sadly have to go through your uni drive unless you are on campus.
From H drive to balena
You can simply copy files across using linux cp commands and the fact that your H drive is called $BUCSHOME on balena. For example, run the command
cp -r folder/with/data $BUCSHOME/destination/folder
to copy a folder with data from balena to your H drive.
From local PC to H drive
You can transfer data to and from University personal drive from your PC by using scp which stands for secure copy and essentially opens up a ssh portal and then closes it again once the files are transferred.
Sending files
Type one of these from within a terminal/kitty/putty on your PC:
scp path/to/directory/f.txt user123@linux.bath.ac.uk:path/to/directory/
scp -r path/to/directory/ user123@linux.bath.ac.uk:path/to/directory/
The first will transfer the file file.txt whereas the second will transfer the whole directory.
Retrieving files
This is the same as above but with the directories reversed in order, for example:
scp user123@linux.bath.ac.uk:path/to/remote/directory/file.txt path/to/local/directory/
Alternatively if you want, you can use something more familiar which is visual. For linux this would be mounting your Uni drive to your PC using a command called sshfs. For Windows you can download an application called WinSCP which looks like it hasn’t changed since 2005 and basically hasn’t but it’s neat and very useful if that’s your thing. A quick google of either of these should give you the information you need. Finally you can use a browser to do this instead if you would like, see here for more details.
Where do I store my files?
So, I will be the first to admit that I have ran long jobs from my $HOME directory. This is bad. It will work but it is faster and better apparently to run your jobs in the $SCRATCH. It’ll be tedious at first but getting used to using simple commands like cp and \verb!ls! will be really helpful. Make sure to save all of your results rather than just plotting graphs at the end, you never know when you might want to alter figures and need the original data again. I would recommend just saving your data and maybe copying it back to your PC to visualise the results. See my guide on how to layout and use a python package for a small research project for more detailed recommendations on how to handle results and plotting for Python specifically.
Jobscripts
The thing you run is the jobscript using the command
sbatch <name of jobscript>.slm
Below I’ve put in an example jobscript that has been adapted from the one given in the scientific computing module. Make sure you are in the directory of the jobscript when you call it and that the program you want to run is in the same folder or that you have specified where it is in the jobscript.
#!/bin/bash
#
# Specify the queue
#SBATCH
# Choose name of job (name of executable)
#SBATCH --job-name = <name of script>.py
# Choose the number of nodes & processors per node:
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#
# Choose the time your code will need at most:
#SBATCH --time=05:00:00
# Add any module which need to be loaded here
module load python3
echo Running on host `hostname`
echo "Starting job..."
# Run the python script $SLUTM_JOB_NAME
# If your script is in MATLAB obviously this needs to be changed
python3 $SLURM_JOB_NAME
The important bits to check are the job name and the time. For ease I have just made the job name the same as the name of the script you want to run. I’ll explain later what the job name is actually used for. Balena allows you free, unlimited usage for jobs which are under 6 hours long so make sure you know approximately how long it will take and change the time given accordingly. Nothing will happen if you overestimate how long you need but it could be helpful for other users to know when their jobs will start if balena is busy and puts your job in ahead of theirs.
If you’re using MATLAB and the paralellisation package thing that I know nothing about, you’ll want to change the number of tasks per node to the maximum of 16 I imagine. Otherwise, unless you’ve done clever things with the MPI4pi or multiprocessing packages in python (which I would highly recommend you learn about if you need to run long programs a lot because this could increase your speed by 16 times or more) then you will probably just be using the one processor and so don’t need to change anything.
Checking Jobs
You will get an output file with the extension .out telling you everything that happens in the job that would normally printed to the screen if you ran it from a terminal.
To check your progress or see if the job has started you can find your jobs by typing
squeue -u user123
Jeremy Worsfold
PhD in Applied Mathematics and Collective Behaviour
My research interests include Collective Behaviour, speficially swarming models and interacting particles Systems. I also have interests in Reinforcement Learning and Scientific Computing.